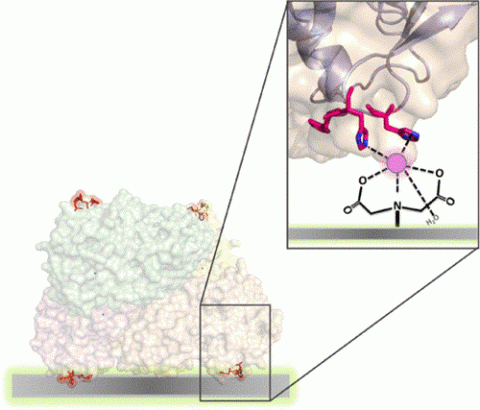
Enzyme immobilization is a key enabling technology for a myriad of industrial applications, yet immobilization science is still too empirical to reach highly active and robust heterogeneous biocatalysts through a general approach. Conventional protein immobilization methods lack control over how enzymes are oriented on solid carriers, resulting in negative conformational changes that drive enzyme deactivation. Site-selective enzyme immobilization through peptide tags and protein domains addresses the orientation issue, but this approach limits the possible orientations to the N- and C-termini of the target enzyme. In this work, we engineer the surface of two model dehydrogenases to introduce histidine clusters into flexible regions not involved in catalysis, through which immobilization is driven. By varying the position and the histidine density of the clusters, we create a small library of enzyme variants to be immobilized on different carriers functionalized with different densities of various metal chelates (Co2+, Cu2+, Ni2+, and Fe3+). We first demonstrate that His-clusters can be as efficient as the conventional His-tags in immobilizing enzymes, recovering even more activity and gaining stability against some denaturing agents. Furthermore, we find that the enzyme orientation as well as the type and density of the metal chelates affect the immobilization parameters (immobilization yield and recovered activity) and the stability of the immobilized enzymes. According to proteomic studies, His-clusters enable a different enzyme orientation as compared to His-tag. Finally, these oriented heterogeneous biocatalysts are implemented in batch reactions, demonstrating that the stability achieved by an optimized orientation translates into increased operational stability.